
INTRODUCTION
Retinitis pigmentosa (RP) is the most common retinal dystrophy [1]. The prevalence of RP is estimated at approximately 1:4000 individuals [2]. A hallmark feature of RP is the degeneration of photoreceptors [3]. The classic triad of signs observed during fundus examination includes bone-spicule pigment deposits in the mid-periphery of the retina – which progressively spread anteriorly and posteriorly while becoming more saturated over time – retinal arteriolar attenuation, and pallor of the optic disc. Macular atrophy, cellophane maculopathy, and macular edema may also occur [4, 5]. RP occurs in various forms that differ in both clinical presentation and inheritance patterns. It may be inherited in an autosomal dominant (AD, 30–40%), autosomal recessive (AR, 50–60%), or X-chromosome-linked manner (5–15%), and it may also occur sporadically [1]. Mitochondrial or digenic inheritance of RP is very rare [2]. The inheritance pattern correlates with disease severity, with AD forms typically showing the mildest course and X-linked forms the most severe [1]. RP is typically confined to the eye; however, there are syndromic forms that involve abnormalities in other systems and organs, as well as systemic forms where retinal disease is secondary to a systemic pathology [2, 6].
Subjective symptoms of RP include gradual loss of peripheral vision, initially manifesting as a ring scotoma and later progressing to tunnel vision. Impaired dark adaptation, night blindness, and photophobia in later stages (when the macula is involved) are also observed. Visual acuity is usually preserved in early stages but declines over time [1–6].
“Leopard skin” retinopathy may appear in several conditions with a clinical presentation similar to RP. “Leopard skin” retinopathy can be caused by medications or systemic diseases, such as arterial hypertension, post-organ transplantation (kidney, heart, liver), immunosuppression, systemic corticosteroids, leukemia, bilateral diffuse uveal melanocytic proliferation, idiopathic uveal effusion syndrome, chronic central serous chorioretinopathy, Refsum disease, β-thalassemia, argyrosis, large cell lymphoma, neonatal adrenoleukodystrophy, Walker-Warburg syndrome, acute posterior multifocal placoid pigment epitheliopathy, and primary intraocular lymphoma. The mechanism of this disorder remains unclear, but a connection between circulatory disturbances at the choroidal level and the development of “leopard skin” retinopathy is suspected [7–11].
Subjective symptoms reported by patients diagnosed with “leopard skin” retinopathy include, among others, blurred vision, gradual unilateral or bilateral deterioration of visual acuity, distorted central vision, difficulty with dark adaptation, metamorphopsia, and eye strain [7–11].
Treatment options for RP are limited. With the exception of gene therapy using voretigene neparvovec-rzyl (Luxturna, Spark Therapeutics) – approved in 2017 by the U.S. Food and Drug Administration and subsequently in 2018 by the European Medicines Agency, where an adeno-associated virus delivers the RPE65 gene to retinal cells – there are no available treatments that halt RP progression or restore vision [12]. Research on additional gene therapies is ongoing [13]. Some studies suggest a correlation between disease progression in certain forms of RP and exposure to sunlight; therefore, patients are advised to wear sunglasses when outdoors. Vita-min A supplementation may be considered, and in patients who additionally present with cystoid macular edema – oral carbonic anhydrase inhibitors, such as acetazolamide, may be used. Other treatment methods, such as transplantation of the photoreceptor layer or the entire retina, are currently being tested in animal models and, less frequently, in humans. To date, however, these have not brought a breakthrough in the treatment of RP in humans [1–3, 5, 6].
No standard treatment for “leopard skin” retinopathy has been described. Whenever possible, therapy is directed toward the ophthalmic or systemic treatment of the underlying diseases [10].
The aim of this paper is to present the diagnostic difficulties in differentiating the hereditary form of RP from “leopard skin” retinopathy.
CASE REPORT
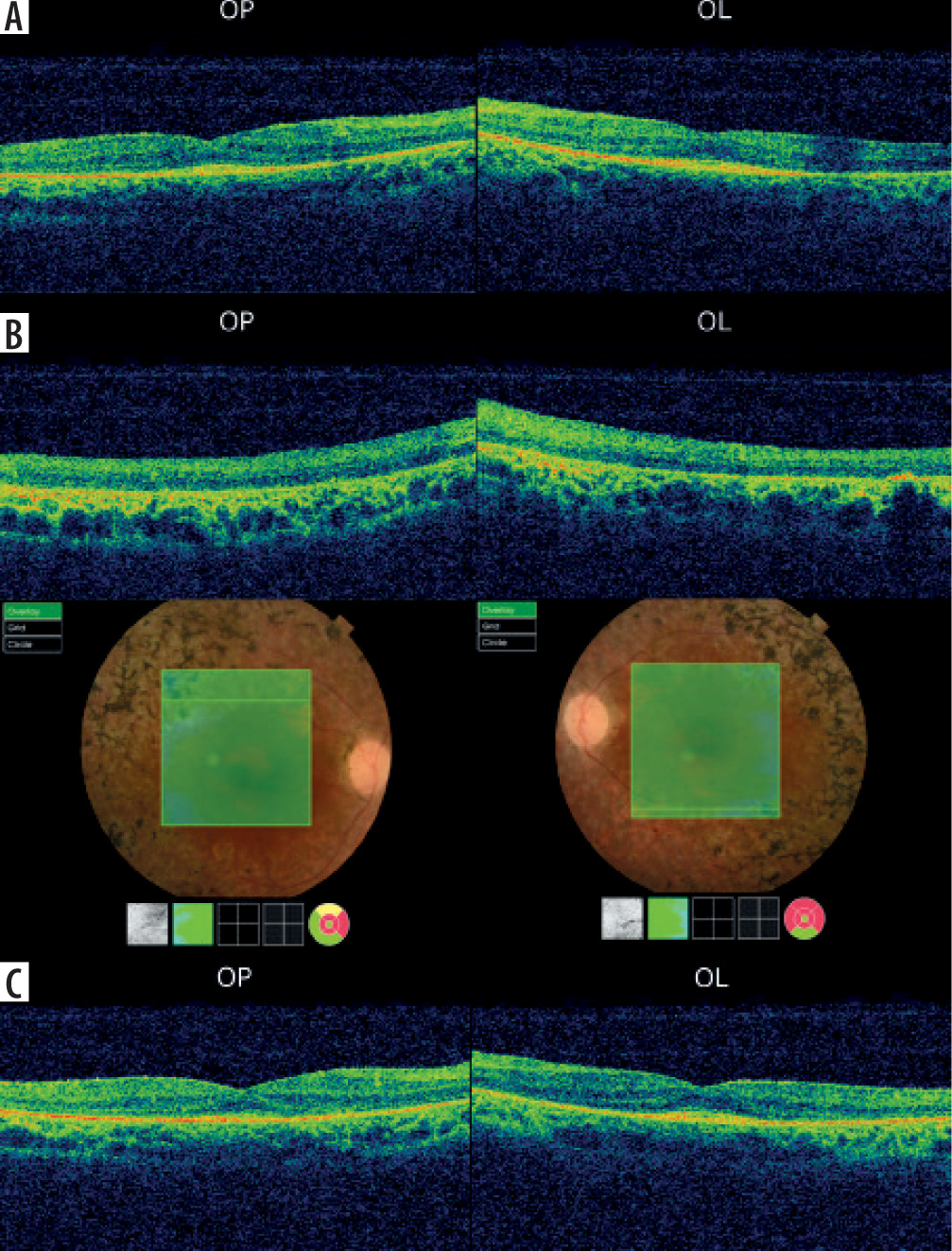
In July 2016, a 34-year-old woman presented to the Clinical Department of Ophthalmology and Ocular Oncology at the University Hospital in Krakow for an ophthalmic examination, including fundus assessment, prior to a scheduled kidney transplant due to stage 5 chronic kidney disease. The patient had no history of prior ophthalmic treatment and denied any visual symptoms. One year earlier, she had been diagnosed with chronic renal failure secondary to hypertensive nephropathy, managed with repeated hemodialysis and peritoneal dialysis. Her medical history also included essential hypertension, secondary hyperparathyroidism, anemia of chronic disease, nephrolithiasis of the left kidney and ureters, and cholelithiasis. The family history for ophthalmic diseases was negative. Physical examination revealed: best corrected visual acuity (BCVA) for distance in the right eye (OD, oculus dexter) and left eye (OS, oculus sinister): 0.6; BCVA for near vision in both eyes: 0.5; intraocular pressure measured by non-contact tonometry was: OD – 14 mmHg, OS – 15 mmHg. Examination of the anterior segment of both eyes revealed no abnormalities. Fundus examination of both eyes revealed pale optic discs, significantly attenuated retinal arterioles, and bone-spicule deposits in the mid-periphery. Kinetic visual field testing (Carl Zeiss Jena kinetic perimeter) showed bilateral, symmetrical constriction of the visual field (OD > OS) (Figure 1A). Macular optical coherence tomography (OCT) (Figure 2A) and a scan through the bone-spicule deposits were performed (Figure 2B). RP was suspected, and genetic testing was recommended; however, the patient declined due to high cost. Fluorescein angiography was not possible due to end-stage renal disease.
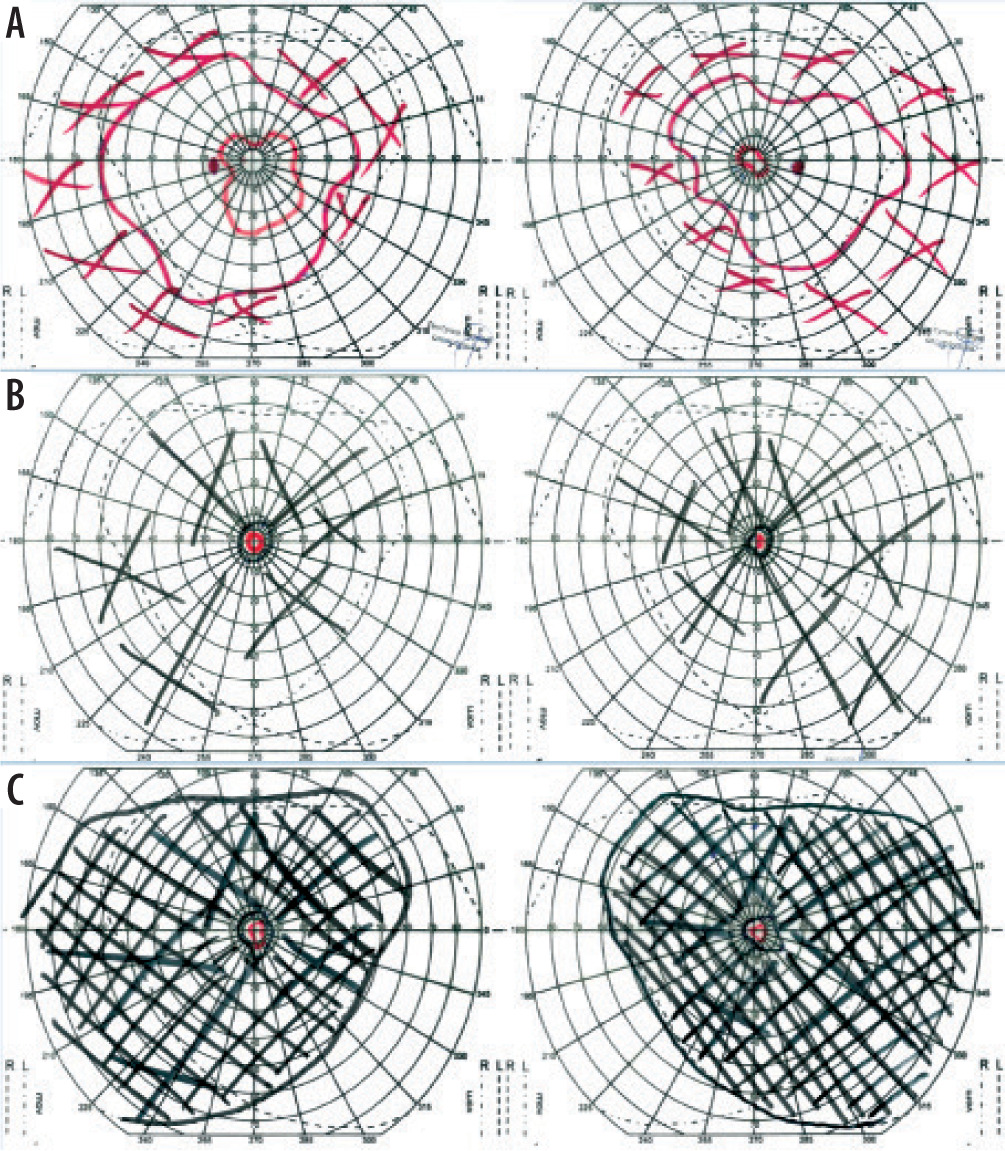
Figure 2
Optical coherence tomography of the right and left eyes: A) maculas and B) bone spicules in 2016; C) maculas in 2020
One year later, the patient underwent deceased-donor kidney transplantation. A triple-drug immunosuppressive regimen was initiated, consisting of tacrolimus, mycophenolate mofetil, and glucocorticosteroids (initially prednisone, followed by prednisolone).
The patient presented for a follow-up ophthalmic examination 6 months after the kidney transplantation. The BCVA for distance and near remained unchanged, despite the development of a slight posterior subcapsular cataract. Fundus examination of both eyes revealed the presence of abnormalities described during the patient’s first visit, as well as macular atrophy, retinal thinning, and more numerous bone spicules throughout the mid-periphery of the eyes.
The next follow-up visit took place in May 2019. The BCVA and anterior segment examination results in both eyes were comparable to those obtained during the previous visit. During the fundus examination, it was observed that the bone spicules reached the optic nerve discs and crossed the vascular arches toward the center. Kinetic visual field test (Carl Zeiss Jena kinetic perimeter) revealed a massive constriction of the visual field in both eyes (OD and OS), compared to the 2016 examination (Figure 1B).
In May 2019, the patient underwent a full-field flash electroretinography using the EP-1000 device (Tomey, Japan) and DTL electrodes. The results showed extinguished retinal activity under scotopic (dark-adapted) conditions and nearly extinguished activity under photopic (light-adapted) conditions. Due to the atypical fundus appearance, the absence of a family history, and the lack of early-onset night blindness, a suspicion of “leopard skin” retinopathy secondary to systemic diseases was raised. Retinopathy of this type has been described in patients undergoing systemic steroid therapy, those with arterial hypertension, organ transplant recipients, and patients with dysproteinemia, occurring as a result of circulatory disturbances at the choroidal level.
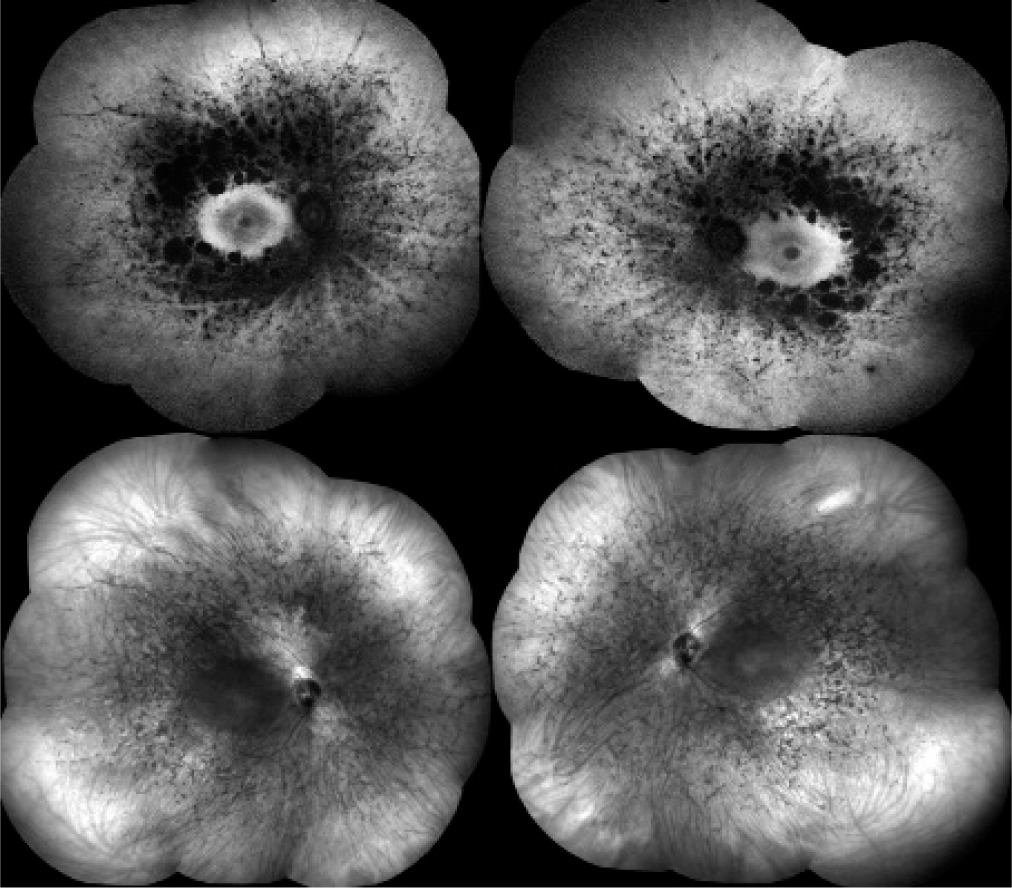
The patient presented for a subsequent follow-up visit in February 2020. She reported no changes in her vision since the previous appointment. BCVA, the anterior segment, and the fundus of both eyes remained unchanged. The tunnel-like constriction of the visual field was also comparable to the previous result (Figure 1C). Macular OCT was performed (Figure 2C), along with optic nerve disc OCT, color composite (mosaic) photograph, and autofluorescence and infrared fundus photography of both eyes (Figure 3 and 4).
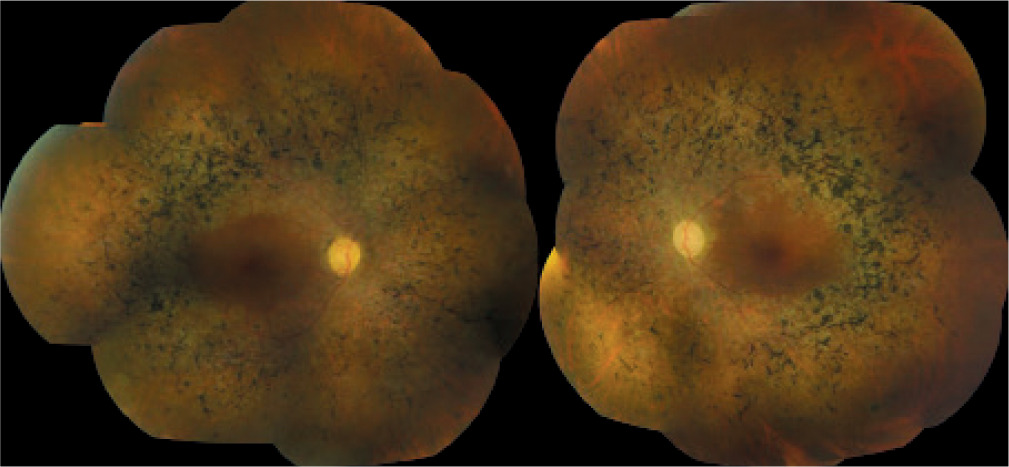
Figure 4
Composite fundus autofluorescence (top) and composite infrared fundus images (bottom) of the right and left eyes
Due to diagnostic challenges, the patient was again advised to undergo an available genetic testing panel for RP with various inheritance patterns. If mutations were detected in the analyzed genes, these tests would confirm the hereditary nature of the disorder. However, the patient again chose not to undergo the tests due to their high cost.
DISCUSSION
The patient described above presented typical features of RP in the fundus examination, such as bone spicule-shaped pigment deposits in the mid-periphery of the retina, which spread over time to reach the optic nerve disc and crossed the vascular arches toward the center, along with retinal arteriolar attenuation, optic nerve disc pallor, and macular atrophy. Minimal posterior subcapsular cataract found during the anterior segment examination of both eyes, combined with progressive visual field constriction, further pointed toward a hereditary form of RP [3–5]. Electroretinography is one of the key diagnostic tests for RP [1–6]. However, the results of this test are not specific; extinguished retinal activity can occur in numerous pathological states. In this case, the clinical presentation and a medical history lacking early symptoms of night blindness appear to be decisive, suggesting a suspicion of “leopard skin” retinopathy secondary to systemic diseases. This diagnosis may also be supported by the patient’s history: end-stage renal disease leading to a kidney transplant from a deceased donor, followed by the administration of immunosuppressive drugs and systemic corticosteroid therapy [7–11]. On the other hand, the kidney transplant took place nearly a year after the patient’s first visit to the Krakow Clinic, at which time she already presented typical features of RP. In that case, “leopard skin” retinopathy would have to have been triggered by previously diagnosed systemic conditions: renal failure due to hypertensive nephropathy treated with repeated hemo-dialysis and peritoneal dialysis, primary hypertension, secondary hyperparathyroidism, and anemia associated with chronic diseases [7–11]. At the same time, the rapid progression of peripheral visual field constriction (to tunnel vision) occurred only after the kidney transplant. In previously reported cases of “leopard skin” retinopathy, the association between the development of this disorder and organ transplantation was more evident; that is, the ophthalmic examination performed before transplantation showed no signs of RP, and these features appeared only about a year after the procedure, during immunosuppressive therapy [7]. Therefore, genetic testing is indicated. However, as a standard, only the most common loci are analyzed for mutations, including the OFD1, RP2, and RPGR genes (for X-linked RP), 16 of the 23 genes responsible for AD RP, and 28 of the 36 genes linked to AR RP. Consequently, only a positive result would be conclusive [14]. Even identical mutations within the same gene may result in different clinical manifestations [14, 15]. A negative result would not rule out a hereditary form of RP caused by a mutation in a different, unanalyzed gene. It should be noted that establishing a definitive diagnosis would have limited impact on therapeutic management given the currently available treatment methods. However, identifying the genetic basis of the disorder could influence future family planning decisions. It is also important to acknowledge the high cost of such testing, which is typically borne by the patient, as well as the limited availability of centers offering these evaluations.
CONCLUSIONS
The presented case study of a patient with suspected “leopard skin” retinopathy provides insight into the diagnostic challenges in differentiating this condition from RP. During the diagnostic process, not only ophthalmic symptoms but also systemic diseases, family history, and occupational and environmental exposures should be taken into account. The wide spectrum of syndromic and systemic forms of RP further underscores the need for a comprehensive medical history. Genetic testing may be helpful in establishing the diagnosis; however, it does not currently enable targeted treatment. In patients following organ transplantation and in the course of other systemic diseases, regular ophthalmic examinations are essential, as they may allow for the early detection of visual impairment and potential modification of systemic therapy. There is a vast number of gene mutations associated with the clinical presentation of RP. It is generally accepted that a negative genetic test result does not exclude a hereditary basis of the disease. Conversely, focusing on potential secondary causes of retinopathy provides an opportunity to modify treatment and limit the impact of systemic factors or medications on the retina.







