
INTRODUCTION
Cryptophthalmos, derived from the Greek term for “hidden eye,” was first described by Zehender, who reported a child whose eyes were completely covered by skin [1]. In the complete form of this condition, the eyelid folds fail to develop, resulting in uninterrupted skin extending from the forehead to the cheek [2]. In contrast, incomplete cryptophthalmos presents an underdeveloped eyelid and conjunctival sac. The condition may affect one or both eyes and can occur either on its own or as part of a broader syndrome, most commonly Fraser syndrome – a rare inherited disorder characterized by cryptophthalmos, fused fingers or toes (syndactyly), and abnormalities of the genitourinary tract [3]. Cryptophthalmos is estimated to affect approximately 3 out of every 100,000 people, with no notable differences among ethnic groups [4]. Because the eyelid fold does not develop properly, the eye is abnormally covered by epithelial tissue, often leading to secondary developmental defects such as corneal hypoplasia, microphthalmia/anophthalmia, or grossly disorganized ocular structures [5].
CASE REPORT
A newborn Caucasian girl was presented to our team 10 days after birth. She was born full term via Caesarean section following an uncomplicated pregnancy. During pregnancy, the mother had no history of noticeable illness, trauma, or exposure to toxic agents or radiation. Her parents reported that she was unable to open her eyes. On examination, there were no upper eyelid creases in either eye. Although this feature is unusual in the Caucasian population, the parents did not find it concerning, as the same characteristic was noted in the father. General systemic examination revealed no other congenital deformities. The parents were not consanguineous; they had normal intelligence and had no congenital abnormalities.
The eyelid fissures were extremely short, making it impossible for the child to open her eyes spontaneously. Bilateral eyebrows were present and normal in appearance. When we attempted to examine the globes using eyelid retractors, only mucous tissue was visible (Figure 1). The left palpebral fissure measured 2 mm shorter than the right (6 mm vs. 8 mm). Bilateral symmetrical partial closure of the eyelids was observed temporally.
Figure 1
Examination of the anterior segment 10 days after birth of the child with an eyelid retractor. A) Right eye – palpebral fissure width about 8 mm. B) Left eye palpebral fissure width about 6 mm
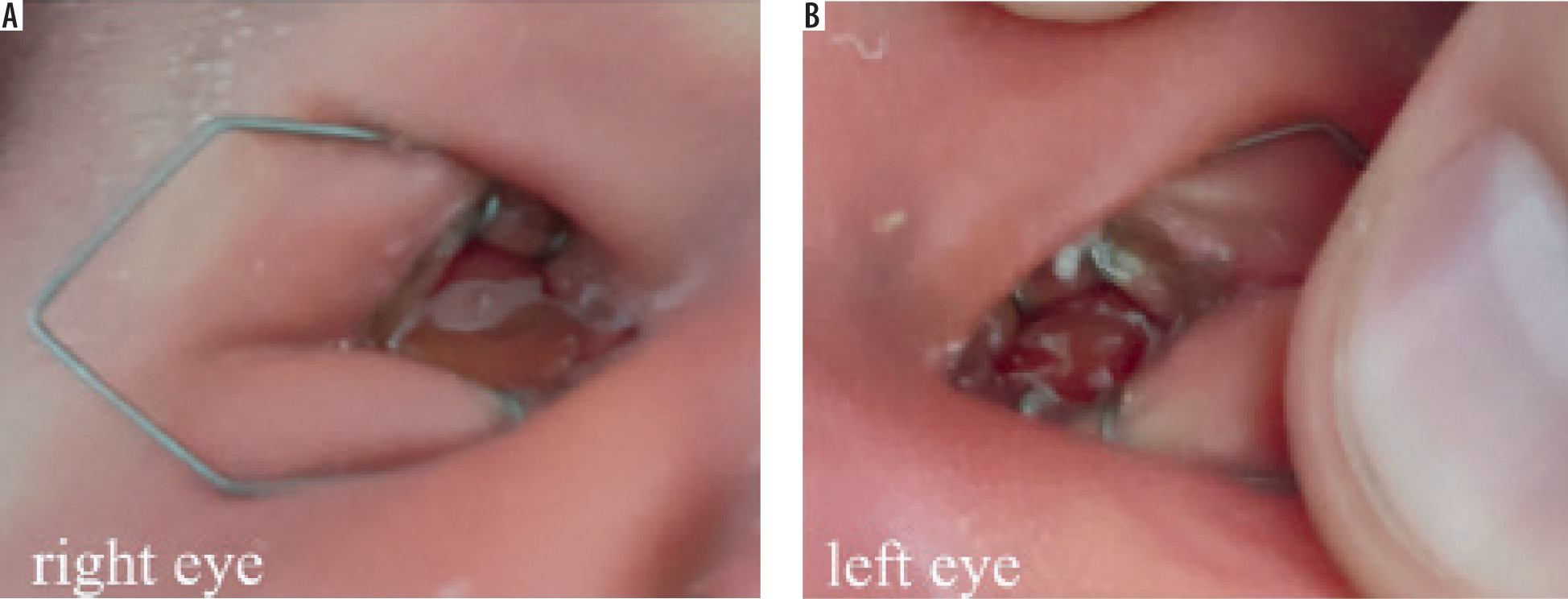
The orbits did not appear empty, and overall, the child gave the impression of being asleep. Eyelashes were present bilaterally, and a small amount of discharge was noted emerging from the conjunctival sac.
During prenatal ultrasound examinations, the evaluating physician did not identify any abnormalities. The mother had a history of a medically induced abortion due to severe morphological anomalies in the fetus that were incompatible with life. The parents did not undergo genetic counseling following that pregnancy termination, as they believed it to be an isolated occurrence.
We decided to perform B-scan ultrasonography (Figure 2) to assess whether ocular structures were present beneath the mucous tissue. In the right eye, we visualized – although not very clearly – some of the normal anatomical components, including the lens, vitreous body, optic nerve, and retina. The left eye demonstrated some identifiable elements of normal anatomy but was significantly smaller than the right.
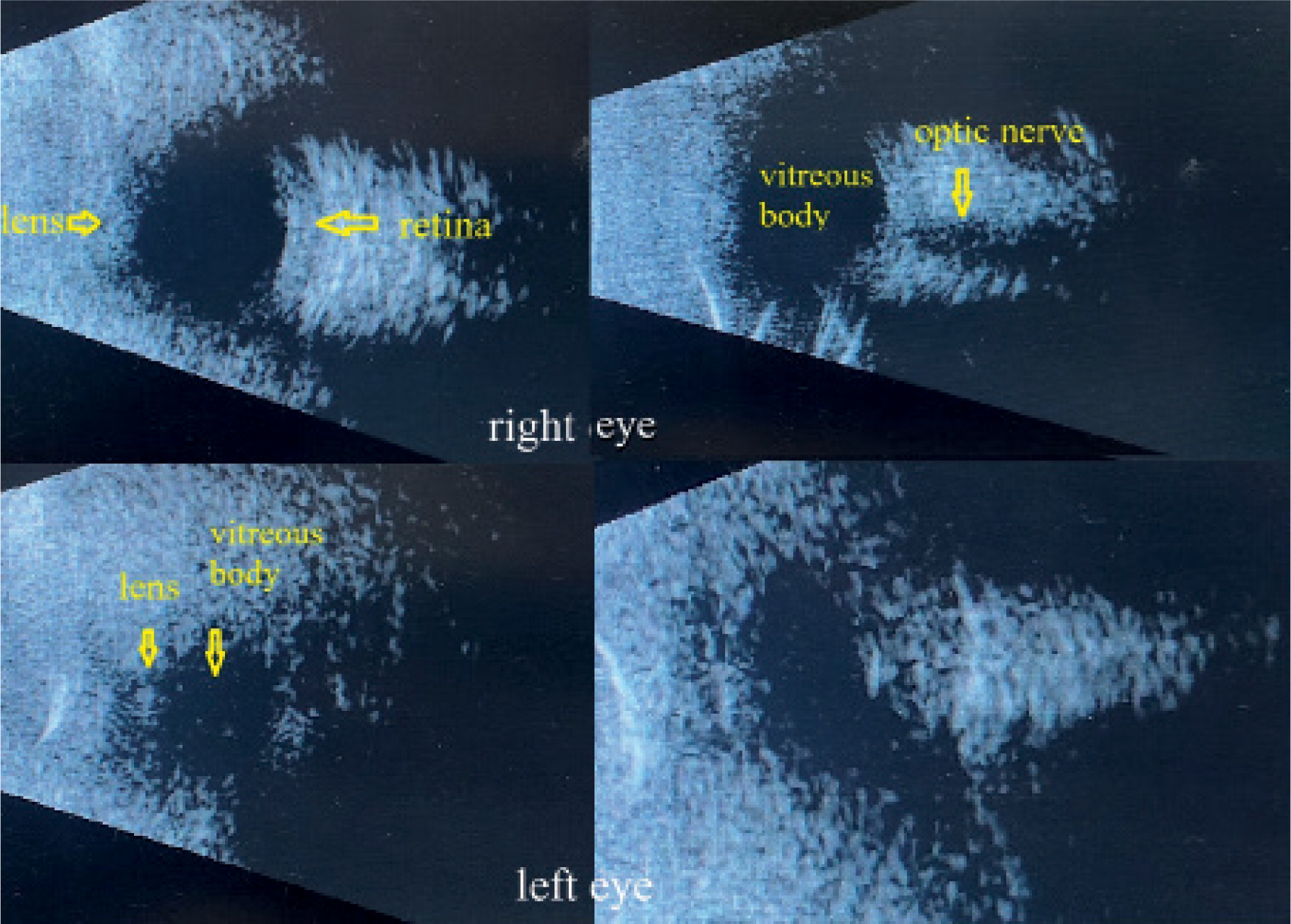
These findings gave us hope that, following reconstructive surgery of the eyelids and fornices and removal of the conjunctiva covering the cornea, it may be possible to achieve some degree of functional vision, at least in the right eye.
We consulted multiple specialists to evaluate the potential for achieving any functional vision, but the opinions were conflicting. Some believed the case represented true anophthalmia, while others suspected the presence of only orbital cysts with no identifiable ocular structures.
Magnetic resonance imaging (MRI) with contrast was performed when the child was approximately two months old to better visualize the ocular structures and assist in making a definitive diagnosis (Figure 3).
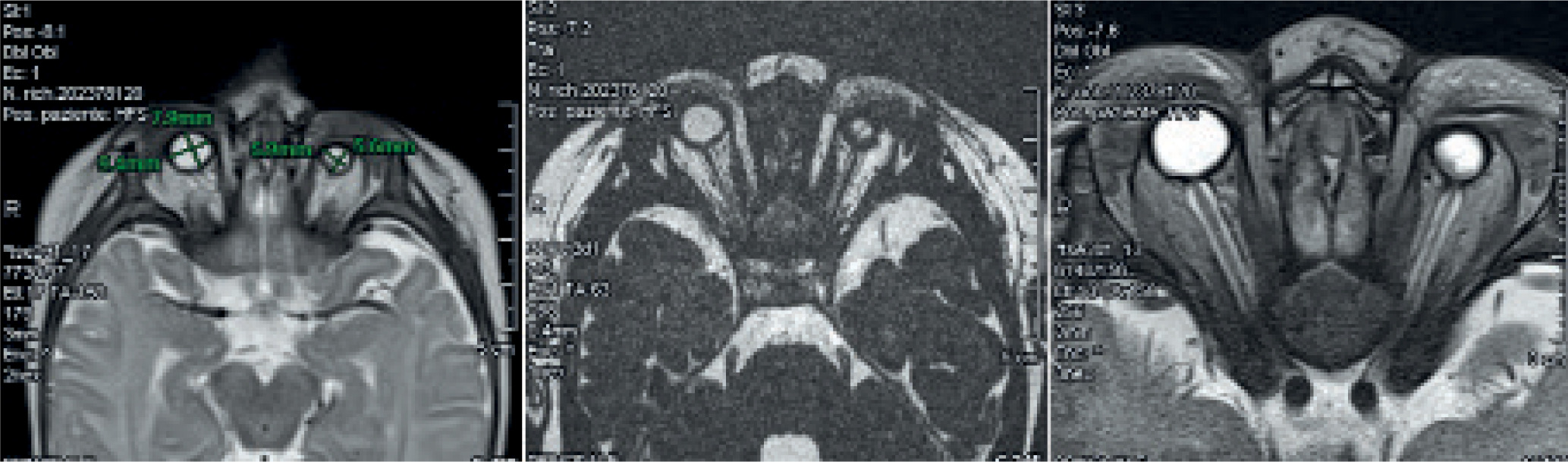
The detailed study of the orbital regions confirmed the presence of bilateral microphthalmia, with a marked reduction in globe size, more pronounced on the left side. The left globe measured approximately 6 × 5.5 mm, while the right measured about 9.5 × 8 mm.
Unfortunately, the optic nerves were thin and not clearly distinguishable bilaterally. The left optic nerve appeared more attenuated than the right. The extraocular muscles, particularly in the posterior region of the left globe, were slightly reduced in thickness and showed a mildly inhomogeneous nodular appearance, though without definitive signs of fat infiltration.
There were no focal or significant signal abnormalities in the supratentorial or infratentorial brain parenchyma. Myelination of the posterior limb of the internal capsule was appropriate for age. No abnormalities in diffusion were observed, and there were no signs of acute ischemia or areas of increased signal intensity. The ventricular system and subarachnoid spaces appeared normal in shape, size, and volume. The midline structures were unremarkable.
At ten months of age, the child was re-evaluated. She demonstrated normal physical and mental development for her age. She was lively, playful, and made visible attempts to open her eyes (Figure 4). She appeared to respond to light sources, showing interest in them.
Figure 4
At ten months, the child attempts to open her eyelids – note the lack of eyelid crease, epicanthus inversus, and short palpebral fissure
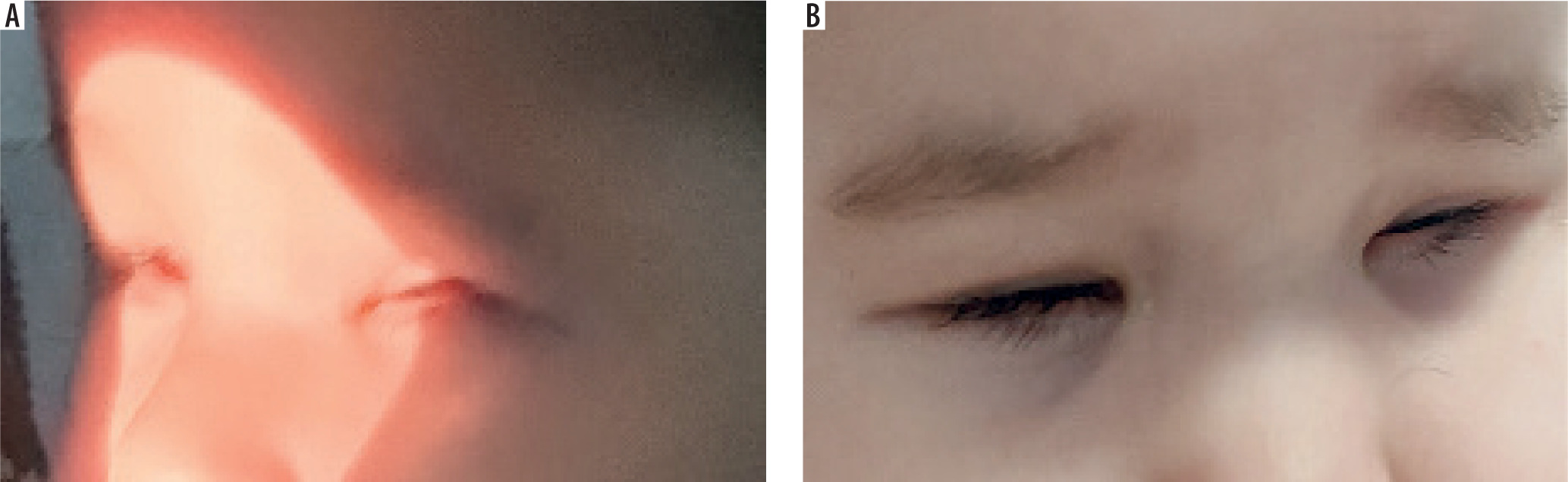
There were still no visible upper eyelid creases. The eyelid fissures were very short, with prominent epicanthus inversus bilaterally and telecanthus.
A chromosomal microarray analysis revealed no pathogenic copy number variants. A targeted gene panel including FRAS1, FREM2, and GRIP1 was recommended to evaluate for Fraser syndrome or other ocular malformation-related genes; however, this testing has not yet been performed due to lim- ited availability and high cost in our country.
DISCUSSION
The differential diagnosis of this clinical case was a challenging one. First, we suspected true anophthalmia due to the absence of recognizable globe structures. Before the MRI examination, some of the consulting specialists suspected ocular cysts. Another potential diagnosis was blepharophimosis syndrome [6] due to the small palpebral fissure, epicanthus inversus, and telecanthus of the patient. However, in this syndrome, the eyes could be observed under the ptotic eyelids. Micro-blepharon [7] was also considered, but in this diagnosis lagophthalmos is usually present, not inability to open the eyelid. The difficulties in opening the eyelids, shortened eyelid fissure, and lack of visible ocular structures covered by mucous tissue, in combination with microphthalmic eyes, gave us confidence that this was a case of incomplete cryptophthalmos.
Prenatal diagnosis is neither frequent nor easy and relies on precise, high-quality ultrasonography [8]. Current ante-natal ultrasound protocols for imaging of the fetal eye are inconsistent and inadequate to screen for the spectrum of ocular malformations, and there are no clear guidelines on detection of these rare abnormalities. This case also underscores the limitations of routine prenatal ultrasonography, which in this patient failed to detect any ocular abnormality during the antenatal period. While high-resolution fetal MRI can improve detection, it is not routinely employed unless there are other anomalies.
The primary aim of surgery in cryptophthalmos is to improve appearance and, when possible, preserve or restore visual function [9]. However, successful cases of visual rehabilitation in these patients are uncommon. For example, Zhang et al. [4] described a patient with complete cryptophthalmos who achieved hand motion vision following ocular surface reconstruction. Similarly, Dibben et al. [10] reported a case of incomplete cryptophthalmos where surgical repair resulted in visual acuity of 20/200. In our case, both optic nerves were hypoplastic, making the chances of recovering any usable vision very poor. When there is no visual potential and the eye does not cause pain, surgical intervention is typically postponed until the child is older, so that more tissue can be used for reconstruction [11]. Reconstructive procedures may include enucleation, creation of conjunctival fornix, reconstruction of the eyelids using local skin flaps and amniotic membrane, and placement of an ocular prosthesis.
Although cryptophthalmos, as a part of Fraser syndrome, has been reported many times, isolated cryptophthalmos without systemic associations is very rare [12]. Bilateral cryptophthalmos with microphthalmia [13], as observed in our patient, is associated with a higher likelihood of syndromic involvement or genetic mutations compared to unilateral cases. Even though chromosomal microarray analysis excluded major chromosomal anomalies, further molecular testing with a targeted gene panel was advised. The family’s history of a previous pregnancy terminated for severe fetal malformations raises the possibility of an unrecognized syndromic or genetic etiology despite the absence of definitive findings so far. Comprehensive genetic evaluation remains an important consideration and may assist in recurrence risk counseling. Unfortunately, such testing remains inaccessible in many regions because of high cost and limited laboratory availability, underscoring the need for broader access to advanced genetic diagnostics in rare congenital disorders.
CONCLUSIONS
This report highlights the importance of a multidisciplinary approach, combining pediatric ophthalmology, radiology, genetics, and reconstructive surgery to achieve accurate diagnosis and guide management in complex congenital eye anomalies. Ongoing follow-up is critical to monitor visual development, provide family support, and address the psychosocial impact of visible facial differences.







